
















Robust, predictive post-market surveillance systems that monitor medical device safety after launch, reduce both costs and demands on resources and increase product safety and performance. However, as competition intensifies, could it be time to revisit the approach?
The historical role of post-market surveillance (PMS) has been reactive. It has focused on collating and reporting adverse events, such as device malfunctions or patient injuries, in order to trigger product returns, modifications, exchanges and recalls as necessary. However, as medical device complexity increases, PMS approaches will need to be more rigorous, leading to earlier detection of potential product failures in the field.
Regulatory authorities are starting to emphasize the importance of PMS plans that are based on proactive data gathering and analysis, rather than relying on reactive data gathering once a serious post-market event has been reported. To meet these needs, manufacturers should take a risk-based approach when considering clinical and post-market needs.
The Food and Drug Administration's approach
Under section 522 of the Federal Food, Drug and Cosmetic Act in the Safe Medical Devices Act of 1990 (SMDA), the FDA is authorized to require manufacturers to conduct PMS for certain class II and class III medical devices. These are:
- devices where failure would have serious adverse health consequences
- devices that are implanted within the body for more than one year
- devices intended to be life-sustaining or life-supporting being used outside a facility
- devices expected to have significant use in pediatric populations
Examples include: infusion pumps (class II); implantable pacemakers (class III); and HIV diagnostic tests (class III).
The FDA can require a surveillance period of up to 36 months or longer, and manufacturers are required to comply with an order for PMS within 15 months of receipt
The European Union's approach
The EU Medical Device Directive (MDD) has always required medical device manufacturers to carry out PMS.
Post-market clinical follow up (PMCF) studies, detailed in the MEDDEV 2 .12/2 Rev 2 in January 2012, are required for some devices that carry potential residual risks or need more clarity on long-term clinical performance, for example:
- innovative products
- products that have undergone significant changes
- high risks relating to the product, its anatomical location of use, or the patient population
- severity of disease
- unanswered questions concerning safety or performance
- issues with trial results, long-term safety, performance, adverse events, surveillance data
- emerging safety or performance data
Many manufacturers distributing CE-marked medical devices in the EU have not recognized the need for PMCF, and relied on the requirements in the January 2013 MEDDEV 2.12/1 Rev 8, which outlines the medical device vigilance system. This requires manufacturers to notify the relevant national competent authority about incidents involving a medical device, and any corrective actions taken. These processes are similar to those outlined in the FDA's Medical Device Reporting.
Once the EU Medical Device Regulation (MDR) is in place this will change as the MDR mandates the need for PMS plans and PMCF.
Developing a good post-market surveillance system
Many medical device manufacturers may not have the experience, expertise or resources to create a centralized safety function and proactive or predictive PMS. They may only realize its importance when faced with findings from regulatory authorities and notified bodies. By this point, the pressure to improve the PMS program is on and an accelerated effort to comply may be prohibitively expensive.
Working ahead of time with an experienced third-party such as IQVIA will help to establish a robust and predictive PMS approach by:
- identifying sources of relevant data and prioritizing the impact on product quality
- proposing a central safety repository to assure data collection, aggregation and analysis, revealing signals of potential issues before they occur
- breaking down silos between functional groups and assuring active participation of all constituents, including distributors
- identifying the technology needs to support the PMS as designed
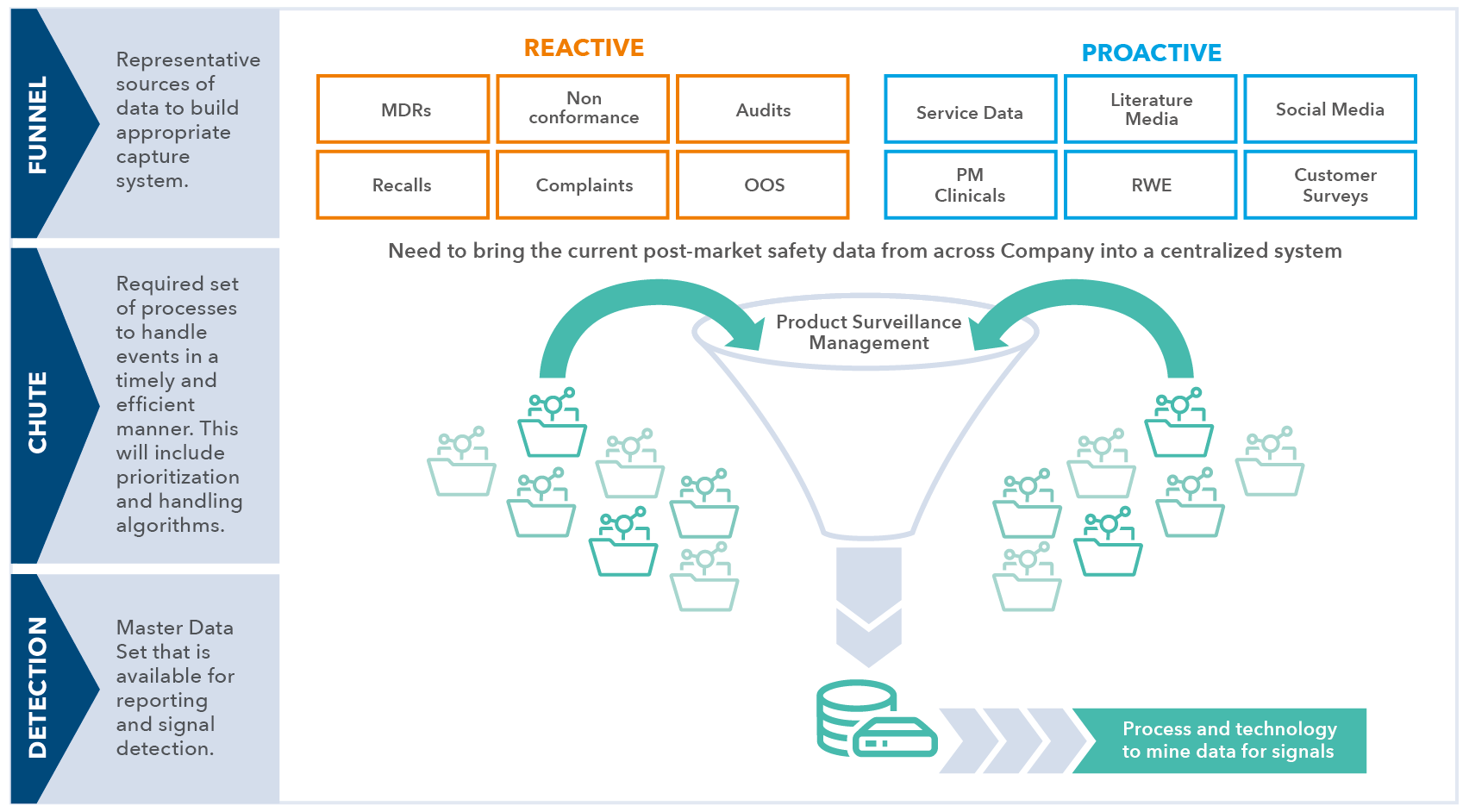
Many medical device manufacturers struggle with the concept of a centralized safety function and a proactive or predictive PMS system. IQVIA has the experience, vision and systems to support manufacturers in identifying the needs and finding the most relevant solution for their organization.
MedTech companies can achieve a competitive edge by early implementation of proactive PMS programs by heading off the tangible costs of down-stream field corrective actions and the very real intangible cost of customer satisfaction.
