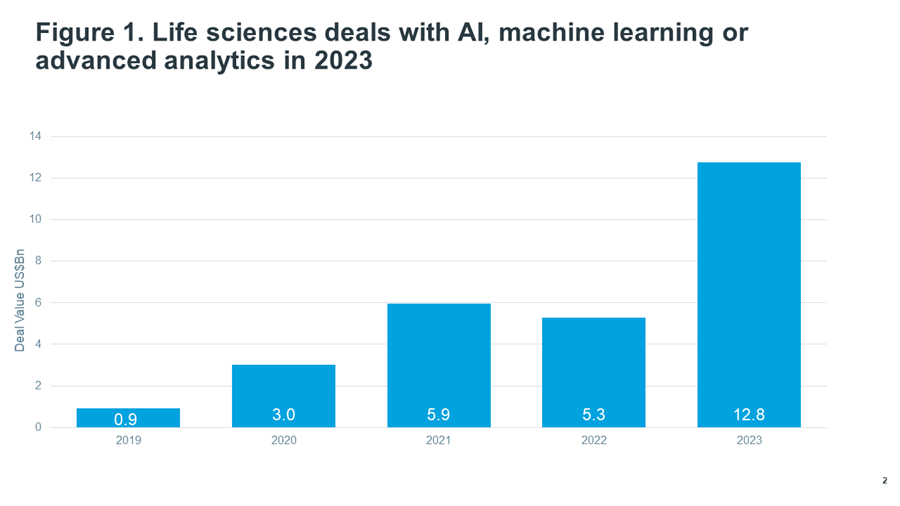
Source: IQVIA Pharma Deals. Dec 2023
A large segment of AI’s use in healthcare would be classified as ‘high-risk’ under the Act and thus subject to multiple requirements if it is developed or deployed within the EU. These requirements will also apply to existing AI systems but only if they undergo ‘significant changes’ to their design after this Act comes into effect.
It should be noted that non-high-risk uses of AI will also require compliance. For example, applications of ‘General Purpose’ AI in business processes may require compliance as detailed below.
What is high-risk AI in healthcare? - The summary
In healthcare, high-risk AI will be that which is used for purposes such as diagnosis, monitoring physiological processes, and treatment decision-making, amongst others. Most of these uses are also software classed as a Medical Device requiring ‘conformity’ assessment by a regulator. The AI Act uses this classification: all AI used in software requiring this Medical Device conformity assessment are classified in the Act as high-risk AI. The only high-risk uses that fall outside of this are specific uses tagged on the end of the Act in Annex III.
What is high-risk AI in healthcare? - The detail
Firstly, we need to look at how AI is defined in the text:
“…a machine-based system designed to operate with varying levels of autonomy and that may exhibit adaptiveness after deployment and that, for explicit or implicit objectives, infers, from the input it receives, how to generate outputs such as predictions, content, recommendations, or decisions that can influence physical or virtual environments;"
Of this AI, those which meet both of the following conditions are classified as high-risk:
“(a) the AI system is intended to be used as a safety component of a product, or the AI system is itself a product, covered by the Union harmonisation legislation listed in Annex II;
(b) the product whose safety component pursuant to point (a) is the AI system, or the AI system itself as a product, is required to undergo a third-party conformity assessment, with a view to the placing on the market or putting into service of that product pursuant to the Union harmonisation legislation listed in Annex II”
In addition, specific use cases listed in Annex III are also classified as high-risk. Of these use cases, the only health-related ones are biometric categorisation, determining eligibility for healthcare and emergency healthcare patient triage systems. It is unclear exactly which real-world uses will be caught by these definitions.
Of the legislation listed in Annex II, the only ones specific to healthcare are Medical Devices and In-vitro Diagnostics Medical Devices (IVD), which include software as a Medical Device.
Therefore, for AI to be caught by the Medical Device legislation, and consequently be considered ‘high-risk’, it must satisfy the definition of a ‘Medical Device’ under the Medical Device Regulation (MDR (EU) 2017/745). The MDR defines Medical Devices as:
“[software]…intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
• Diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease;
• Diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability;
• Investigation, replacement or modification of the anatomy or of a physiological or pathological process or state;
• Providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations.”
With this, the EU intends that all AI used within Devices (including IVDs) that require conformity assessment will be classified as high-risk. This would be in addition to any that have an AI system that is intended to be used as a safety component, or AI use cases in Annex III.
Within the MDR, those which require a conformity assessment and thus would be classified as ‘high-risk’ AI are all type IIa, IIb and III:
“Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
- death or an irreversible deterioration of a person's state of health, in which case it is in class III; or
- a serious deterioration of a person's state of health or a surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. [Including software for contraceptives]
All other software is classified as class I.”
For IVD Medical Devices, the vast majority require conformity assessment and would be classified as high-risk.
In sum, high-risk AI is that which is used within software that is classified as a Medical Device, if this Device is risky enough to require ‘conformity’ assessment by a regulator.
However, there is a conflict between the classification intention of the Commission and the wording: if the AI only forms a minor, non-safety part of a Device/IVD, rather than being a software-based Device itself, then the wording of paragraph (a) above is ambiguous. For example, it is unclear whether AI used to improve the user interface of an insulin pump would mean it is classed as high-risk AI.
Importantly, there are exceptions to the above high-risk classification rules: In addition, certain uses of AI are excluded from the scope of this Act:
- Military, defence or national security purposes
- Scientific research and development
- Free and open-source AI unless they are used as prohibited, general purpose AI or in high-risk AI systems
- AI uses listed in Annex III that do not lead to a “significant risk of harm…because they do not materially influence the decision-making or do not harm those interests substantially".
Exactly what ‘significant risk of harm’ and ‘scientific research and development’ mean is unclear, the latter being of particular importance for the impact of this legislation on medicines development. These are expected to be clarified in forthcoming guidance.
What are the regulatory requirements for high-risk AI?
For providers of high-risk AI, there are a multitude of requirements relating to:
- Data and data governance
- Standards, including accuracy, robustness and cybersecurity
- Technical documentation and record keeping
- Continual compliance:
- Risk management system (RMS), including a fundamental rights impact assessment
- Quality management system (QMS)
- Authorised representatives
- Post-marketing requirements
- Oversight:
- Transparency and provision of information to deployers
- Human oversight
- Regulatory Sandboxes
- Privacy
- Accessibility
- Specific obligations for providers, importers, distributers and users
For providers of ‘General Purpose AI’, there is a set of additional requirements, specifically for those with systematic risks. General purpose AI is defined as:
“An AI model, including when trained with a large amount of data using self-supervision at scale, that displays significant generality and is capable to competently perform a wide range of distinct tasks regardless of the way the model is placed on the market and that can be integrated into a variety of downstream systems or applications. This does not cover AI models that are used before release on the market for research, development and prototyping activities;”
For general purpose AI, ‘codes of practice’ will be developed to help providers be compliant. They must also make sure downstream users have a good understanding of the models so that they can integrate them into other systems in compliance with this and other regulations (in reality, this entails providing a multitude of documentation (Annex (IXb) and Annex (IXa)).
Companies that originally use such GPAI models as a Deployer, but who then augment that model may also be subject to these requirements, having made themselves a Provider of a 'new' GPAI model (in respect of the augmented or modified version). Unfortunately, the AI Act doesn't clarify what level of modification would constitute the creation of a new GPAI model. For example, a company that takes a GPAI Large Language Model, and trains it on new data to predict cancer disease progression in individual patients, and tunes it to do so, may in effect have generated a new GPAI model. Were that to be the case, and the model deployed, the company would then have to adhere to the requirements for GPAI providers, as well as those of high-risk AI detailed above.
Providers of open-source GPAI systems are exempt from certain requirements. Additionally, in the EU’s efforts to recognize regulatory burden, SMEs and microenterprises are subject to fewer regulatory requirements.
These regulatory requirements do not just affect providers of AI: importers, distributers and deployers of AI have requirements too. For example, a healthcare professional or their organisation using AI to advise on treatment decisions would be subject to the regulatory requirements for deployers:
- Human oversight and logging decisions
- Sticking to the appropriate uses pre-defined by the provider
- Ensuring the input data to the AI is relevant
- For Annex III uses, informing the natural persons that they are subject to the use of the high-risk AI system
- Vigilance and reporting of adverse incidents or risks
- Cooperation with authorities, including data sharing.
The requirements of this Act overlap with requirements of other regulations, in particular the MDR. Therefore, freedom is given as to how to comply with different regulations at the same time e.g. complying with this Act’s requirement for a RMS by amending the existing RMS of a Medical Device. However, if there is a conflict in regulatory requirements, it is unclear which requirements will take precedent.
There are also new bodies created - an exciting concept for the EU political class - and along with existing standard-setting bodies, these will develop standards to help comply with this Act. However, there are concerns that the EU body producing the standards set out in this Act relevant to Devices, CENCELEC, may produce EU-specific standards that differ from the global, ISO standards favoured by providers. This would lead to divergence in requirements and force providers to choose one or the other or both standards. On top of this, these EU standards are behind schedule to be ready by the time this Act comes into effect, casting uncertainty on how to comply.
Who will assess them?
For most high-risk AI used in healthcare, compliance will be assessed by the medical device regulators (‘notified bodies’) through the existing conformity assessment procedure, providing these bodies have been authorised by Member States to assess compliance to this Act. This authorisation is another cause for concern as it is unclear how the Commission intends to ensure enough notified bodies are authorised in time, especially given how stretched notified bodies have been to certify Medical Devices under the recent Device Regulation.
As compliance assessment occurs at a single point in time but because AI updates and evolves, this Act helpfully includes the concept of pre-agreed changes not requiring re-assessment.
When will it come into effect?
For prohibited AI, obligations will apply 6 months after the entry into force of this Act, for general purpose AI after 12 months and for high-risk AI after 3 years. For existing high-risk AI used within specific large-scale IT systems (listed in Annex IX) or intended to be used by public authorities, they will apply by the end of 2030 and four years, respectively.
How will it affect my organisation?
Penalties for non-compliance are serious, stretching up to 7% of global turnover.
Organisations should develop an AI governance and compliance strategy, taking advantage of existing risk management processes. These should be based around this Act but also principles of responsible and ethical AI use to ensure they are future-proofed to other AI regulations. Organisations should consider establishing AI regulatory affairs roles to manage AI regulatory risks from this Act and others that will begin coming, globally.
If you have further questions on mapping your organisation’s AI uses, assessing compliance risks to this Act and others, or implementing AI governance and compliance, get in contact.